本特规则
在化学中,本特法则(英语:Bent's rule)描述并解释了分子的中心原子的轨道杂化与取代基的电负性之间的关系。[1][2]亨利·A·本特发表的规则如下:[2]
原子杂化轨道中,指向电正性取代基的杂化轨道带有较强的s轨道特征
分子的化学结构与其性质和反应性密切相关。价键理论提出分子结构是由原子之间的共价键连接决定的,每条键由两个通常轨道杂化而成的原子轨道重叠而成。传统上,p区元素元素被假定为采取严格的spn方式,其中 n 是 1、2 或 3。其中,每条杂化轨道被假定为等价的。这种方法通常能给出较好的预测和解释,但在解释杂化轨道中所含s成分和p成分的比例不完全相等的不等性杂化时,需要改进。本特规则提供了关于如何构建这些杂化轨道的定性估计。[3]
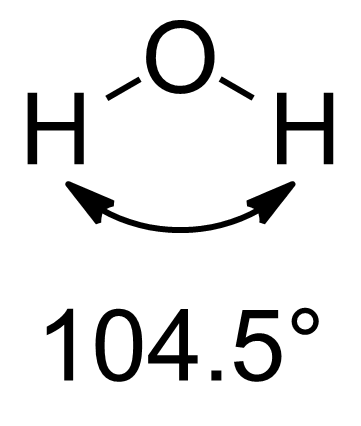
本特规则指的是,在分子中与多个取代基键合的中心原子会发生杂化,其中指向电正性取代基的杂化轨道具有更多的s轨道的特征,而指向电负性取代基的杂化轨道具有更多的p特征的轨道。这样可以更好地预测和解释分子结构和键强度等性质。[4]比如对于水分子而言,其中氧原子的孤对电子可视作连接电正性最大的取代基,其杂化轨道为sp2.3,含有约30%的s轨道和约70%的p轨道。而连接氢原子的成键电子对为~sp4.0,含有约20%的s轨道和约80%的p轨道。最近检查了Bent规则对主族元素之间七十五种键类型的有效性。[5]
应用
本特规则可以被用于解释分子结构和反应性。
键角
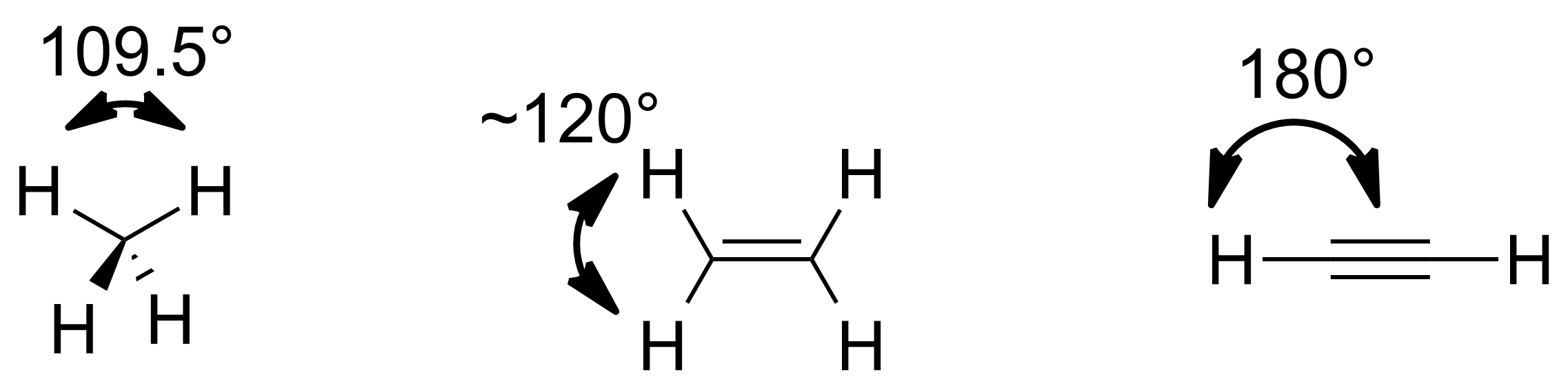
杂化轨道理论解释了为什么甲烷是正四面体结构而乙烯是平面结构,但对于偏移理想结构键角的氨分子和水分子,一般采用VSEPR理论的解释,即孤电子对之间的排斥要大于成键电子对之间的排斥。本特规则提出了另一种解释。首先考虑甲烷、乙烯和乙炔的键角,三者中碳的杂化方式分别是sp3、sp2和sp杂化,对应的键角为约109.5°, 约120°and 180°。可见当杂化轨道中含有较高比例的p轨道时,会造成较小的键角。This result can be made rigorous and quantitative as Coulson's theorem (see Formal theory section below).
{kind=link}
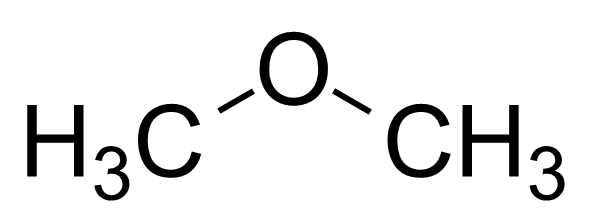
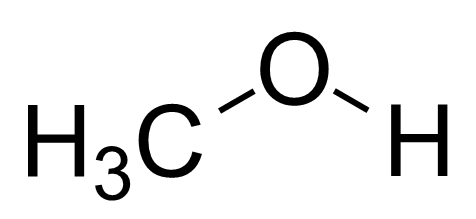
本特利用p轨道的比例和键角的关系,比较了下列分子的键角。考虑到甲基的电负性<H<F,连接甲基的杂化轨道中s轨道的比例应该最多,所以下面分子的键角应该是递减的,这与实验结果一致[2]
| 分子种类 | 取代基之间键角 |
|---|---|
| 二甲醚 |
111° |
| 甲醇 |
107-109° |
| 水 |
104.5° |
| 二氟化氧 |
103.8° |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
本特规则认为孤电子对相当于连着很高电正性的取代基。所以对于水分子,孤电子对含有较高比例的s轨道,而连着氢的杂化轨道含有较高比例的p轨道,于是成键电子对之间的键角较小。氨分子的情况类似,其连着氢的杂化轨道的形式为(~sp3.4,约含23% s轨道,而孤电子对为~sp2.1含有大概32%的s。造成氨分子的键角为107.0。因此可以预测NH3的键角大于NF3的键角,这与VSEPR理论的预测相反,但与实测一致。[6]
键长
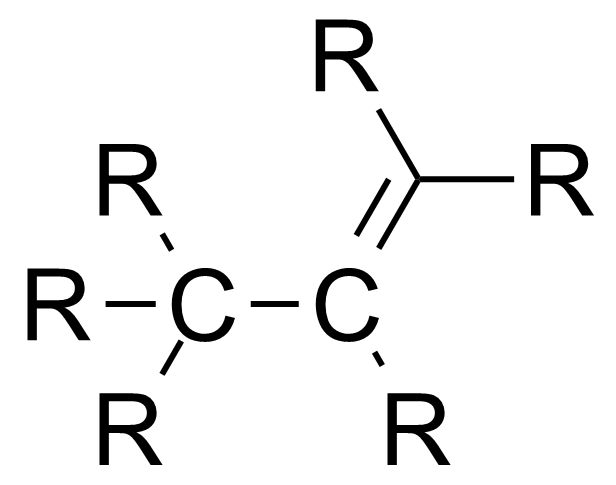
与键角类似,键长与成键原子的杂化轨道有关,s轨道的比例越高,键长越短。[2]
| 分子种类 | 平均碳-碳键长 |
|---|---|
| 1.54 Å | |
| 1.50 Å | |
| 1.46 Å |
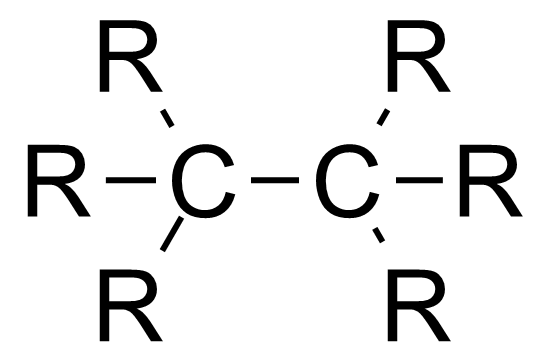{kind=link}
{kind=link}
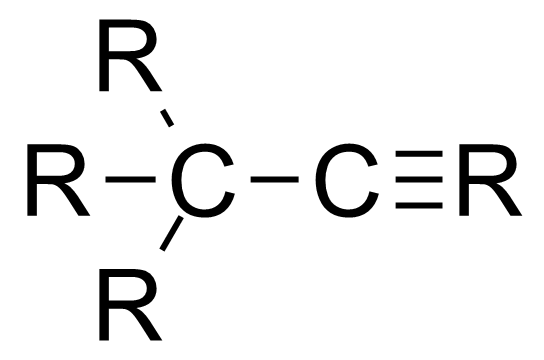{kind=link}
从下图中可以看出,当高电负性的F原子不断取代氢原子时,C-F键的杂化轨道中s的比例在增加,p轨道的比例在降低,所以键长逐渐变短。
| 分子种类 | 平均碳-氟键长 |
|---|---|
| 一氟甲烷 |
1.388 Å |
| 二氟甲烷 |
1.358 Å |
| 三氟甲烷 |
1.329 Å |
| 四氟甲烷 |
1.323 Å |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
同样的趋势也可以鉴于甲烷的四种氯代物。但由于氯的电负性要小于氟,键长差别变得比较小[2]
| 分子种类 | 平均碳-氯键长 |
|---|---|
| 一氯甲烷 |
1.783 Å |
| 二氯甲烷 |
1.772 Å |
| 三氯甲烷 |
1.767 Å |
| 四氯甲烷 |
1.766 Å |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
诱导效应
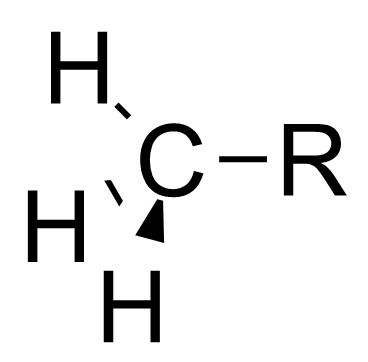
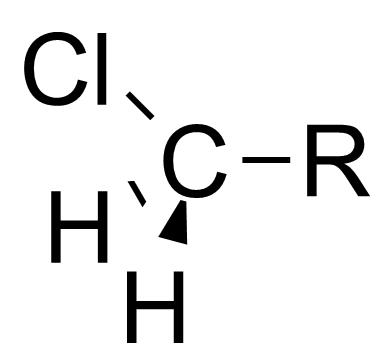
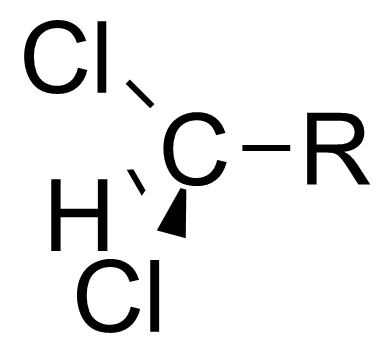
本特规则也可以解释诱导效应。[7]诱导效应指的是部分电荷可以沿着相连的共价单键进行传递,比如当连在中心碳原子上的官能团变得电负性更高时,中心碳原子会变得更加拉电子。本特规则认为,当官能团的电负性增大时,官能团和中心碳原子之间的杂化轨道中p轨道的比例较高,造成了中心碳原子和烃基之间的杂化轨道s轨道的比例较高。因为s轨道比p轨道更接近原子核,所以烃基上的电子云密度更接近于中心碳原子。 这使得中心碳原子更显示出拉电子的诱导效应[8] [9]
| Substituent | 极性取代基常数 (较大值表示较强的拉电子能力) |
|---|---|
| 叔丁基 |
−0.30 |
| 甲基 |
0.00 |
| 氯甲基 |
1.05 |
| 二氯甲基 |
1.94 |
| 三氯甲基 |
2.65 |
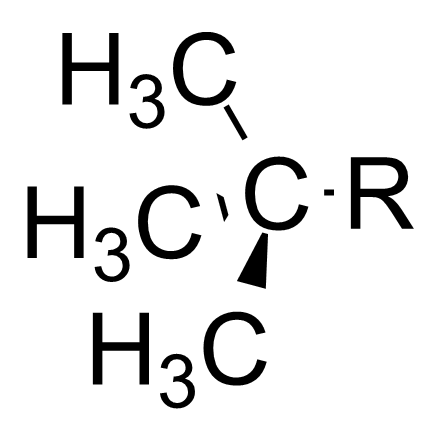{kind=link}
{kind=link}
{kind=link}
{kind=link}
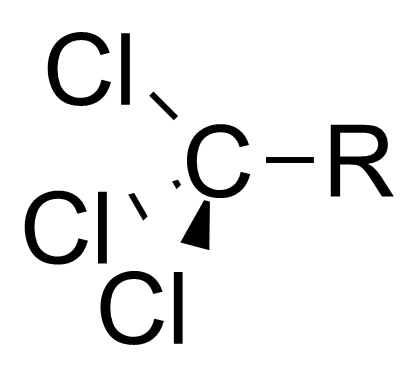{kind=link}
参考文献
- ^ Weinhold, F.; Landis, C. L., Valency and Bonding: A Natural Donor-Acceptor Perspective 1st, Cambridge: Cambridge University Press, 2005, ISBN 978-0-521-83128-4
- ^ 2.0 2.1 2.2 2.3 2.4 Bent, H. A., An appraisal of valence-bond structures and hybridization in compounds of the first-row elements, Chem. Rev., 1961, 61 (3): 275–311, doi:10.1021/cr60211a005
- ^ Foster, J. P.; Weinhold, F., Natural hybrid orbitals, J. Am. Chem. Soc., 1980, 102 (24): 7211–7218, doi:10.1021/ja00544a007
- ^ Alabugin, I. V.; Bresch, S.; Gomes, G. P. Orbital Hybridization: a Key Electronic Factor in Control of Structure and Reactivity. J. Phys. Org. Chem. 2015, 28 (2): 147–162. doi:10.1002/poc.3382.
- ^ Alabugin, I. V.; Bresch, S.; Manoharan, M. Hybridization Trends for Main Group Elements and Expanding the Bent's Rule Beyond Carbon: More than Electronegativity. J. Phys. Chem. A. 2014, 118 (20): 3663–3677. Bibcode:2014JPCA..118.3663A. PMID 24773162. doi:10.1021/jp502472u .
- ^ Weinhold, F.; Landis, Clark R. Discovering Chemistry with Natural Bond Orbitals. Hoboken, N.J.: Wiley. 2012: 67–68. ISBN 9781118119969.
- ^ Bent, H. A., Distribution of atomic s character in molecules and its chemical implications, J. Chem. Educ., 1960, 37 (12): 616–624, Bibcode:1960JChEd..37..616B, doi:10.1021/ed037p616
- ^ 引用错误:没有为名为
Walsh的参考文献提供内容 - ^ Taft Jr., R. W., Concerning the Electron—Withdrawing Power and Electronegativity of Groups, J. Chem. Phys., 1957, 26 (1): 93–96, Bibcode:1957JChPh..26...93T, doi:10.1063/1.1743270